AGENTS OF TIME
Human biology is finite and yet so incredibly complex. We are constantly surprised by new components and systems being discovered. So far we relied on ‘Hallmarks of Aging’ as a reference for what all changes in us with aging but there have been a flood of papers recently that go much more deeper or upstream, closer to the source of these macro changes. They are giving us another perspective on the changes occurring in us with aging.
All of them not only sit very well with but also embellish what I shared in my two previous posts: Headwaters and Autologous Regulation.
So let’s go over some of them here:
Cell-free chromatin particles from dead cells accumulate in our plasma and cut DNA in healthy cells making 1 Quadrillion double strand DNA breaks everyday in each of us as we grow older!
https://www.nature.com/articles/s41598-022-21388-w
A Shocking discovery of how shrapnel from exploding dead cells cause Double Strand DNA fractures in healthy cells and a simple oral supplement may protect against that.
Dr. Indraneel Mittra is a brilliant scientist and a cancer surgeon with Tata Memorial Center for cancer research. He worked against all odds for 20 years to confirm, with evidence of 150,000 volunteers, the importance of regular low cost screening for breast and cervical cancers thereby saving lives of millions of women. Recently he was perplexed by the very high occurrence of double strand DNA breaks in our cells: 10 to 50 per cell per day! These DDBs are constantly repaired to maintain the viability of the cell but any error leads to cell death or worse cancer. So he and his team began to investigate the cause. They were surprised by what they found. Every day around 450 billion cells die in our body (our body is a collection of avg 33 trillion cells). This huge number of dead cells leave fragments or debris which circulates in our blood. Phagocytes work relentlessly to ‘eat’ this debris and digest it - clear it. But Dr. Mittra found that cell free chromatin particles released from the dead cell fragments enter healthy cells and cause double strand DNA breaks which basically snaps both the strands of the DNA. He found that the plasma consists of large quantities of these dangerous bullets circulating all across our bodies as we age they inflict dsDNA breaks, activate apoptotic pathways and induce inflammatory cytokines. This could be one of key causes of rising chronic inflammation during aging. Chronic inflammation is a precursor to most of our mortal diseases. Their group has successfully isolated and characterised cfChPs from human serum, which upon EM examination revealed extensive size heterogeneity ranging between ~ 10 and ~ 1000 nm13. They have also reported that blood levels of cfChPs increase with age. No surprise there. This is a BIG discovery! Dr. Mittra hypothesizes that the that lifelong assault on healthy cells by cell- free chromatin particles or cfChPs is the underlying cause of aging. Again a very major discovery if it turns out to be true. I personally do not fully attribute the circulation of these dangerous particles as the cause of aging because I am sure in the young, healthy body they can be more or less cleared by our amazing phagocytes. What happens with aging is that the efficiency of the phagocytic cells goes down which would leave accumulating amounts of this harmful circulating debris. An example of why phagocytes may lose their efficiency is the disruption of mitochondria seen in aging. Phagocytes especially require huge amount of energy provided by the mitochondria. Here is a paper on that: https://www.cuimc.columbia.edu/news/change-mitochondria-critical-clearing-dead-cells
Regardless of whether cfChPs are the cause of aging or result of aging Dr. Mittra also found that just by taking a combination of resveratrol and copper in a particular ratio one can drastically reduce the cfChPs and in his experiment he saw reversal of many key pathologies of aging. Basically this combination which is absorbed from the stomach after an oral pill generated oxygen radicals that degraded cfChPs. I always like to share some way one could intervene against age related damage whenever something was available. Dr. Mittra’s lab found that oxygen radicals that are generated upon oral administration of R–Cu are apparently absorbed from the stomach to have systemic effects in the form of deactivation/eradication of extracellular cfChPs. In that study they have taken advantage of cfChPs deactivating property of R–Cu to investigate whether prolonged administration of R–Cu to aging mice will retard the hallmarks of aging and neurodegeneration. The dose of Resveratrol used in our study was 1 mg/kg, and that of Copper was 0.1 μg/kg, given by oral gavage twice daily. This dose of Copper was 20,000 times less, and that of Resveratrol 5 times less, than those that have been used in pre-clinical studies to investigate their health promoting properties by other investigators.
Using confocal microscopy and antibodies against DNA and histone they detected copious presence of extra-cellular cfChPs in brain of aging mice, and observed that cfChPs were deactivated/eradicated following prolonged oral administration of R–Cu. Deactivation/eradication of cfChPs was associated with down-regulation of multiple biological hallmarks of aging in brain cells. At a systemic level, R–Cu treatment led to significant reduction in blood levels of glucose, cholesterol and C-reactive protein. Such a combination in the ratio discovered by Dr. Mittra can be an inexpensive, non-toxic solution which can be easily incorporated into our daily supplement regimen.
Global loss of Heterochromatin and disorganization of nuclear architecture in aging
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3414389/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7253059/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8673774/
During youth DNA is tightly packed in heterochromatin but in aging we see rising disorganization in the heterochromatin leading to loosely packed DNA and aberrant transcription of silenced genes. We have 200 types of cells but to create a particular type of cell and maintain that identity nearly 80% of the DNA needs to be permanently silenced and made inaccessible in tightly packed heterochromatin. Aging causes global disorganization of chromatin architecture. Disruption of nuclear lamina/INM proteins and decondensation of associated heterochromatin are common features of normative aging. This initially leads to transcription of genes that should not have been transcribed and later leads to loss of cellular identity causing pro inflammatory secretions and loss of function. Nuclear envelope dysfunction leads to alterations in nuclear transport, chromatin organization, and telomere maintenance and are correlated with nuclear protein alterations, namely nucleoporins, nuclear transport factors, lamins, INM proteins, chromatin‐associated factors, histones modifications, and sheltering complex proteins, revealing that NE proteins are essential determinants of aging. There are also morphological changes that are observed of the nucleus. For example in the young the nuclear periphery is smooth but over time, an increasing number of nuclei started to show a rather convoluted nuclear periphery, with multiple folds. As we grow older we see nuclei that showed increasingly abnormal shape and/or extensive stretching and fragmentation.
Lipotoxic accumulation of lipid droplets in cellular nuclear compartments during aging
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10086520/
The authors say: Age‐dependent deterioration of lipid metabolism and nuclear morphology are common features in evolutionary divergent organisms. Nuclear lipid droplet (nLDs) deposition and LMN‐1/LMNA accumulation lead to cellular dysfunction and, subsequently, to tissue homeostasis collapse, with age. Nothing is kept by Nature and evolution without a purpose. Nuclear lipid droplets would have beneficial functions during embryogenesis, development and youthful homeostasis but during aging they begin to accumulate abnormally within the nucleus causing lipotoxic and mechanospatial damage to nuclear compartments and membrane. The authors found that longevity interventions like fasting and inhibition of IGF-1 reduced such lipotoxic burden. They also examined long lived nematodes and found some commonality with longevity mediation. HLH-30 is a crucial regulator of autophagy and it was found to reduce lipid accumulation in the nucleus in both cases. Specifically an enzyme ATLG-1 which is transcriptionally regulated by HLH-30 maintains lipid droplet homeostasis in nuclear compartments. ATLG-1 deficiency is seen in aging cells of normal worms but not in long lived works and is also upregulated during longevity interventions like fasting in normal worms. This interesting research paper shows how we could investigate further upregulation of ATLG-1 during aging by a natural extract called Oridonin: https://www.nature.com/articles/s41419-023-05613-6
Another deficiency they found in patients suffering from metabolic syndrome is deficiency in Phosphatidylethanolamine PE. Supplementation of PE or Oridonin led to restoration of lipid homeostasis by activating LXRa which in turn activated ATLG-1 and another enzyme EPT-1.
The loss of cellular bioelectricity and polarity in aging
https://www.sciencedirect.com/science/article/abs/pii/S1568163712000992
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3896621/
A fundamental need of our cells is bioelectricity otherwise it would shut down just like any mobile phone when batteries run out. Similarly cells in our body progressively loose polarity as we age (or during cancer and disease). Polarity is important for the spatial organization of a cell. We read above what happened when a tight, stable configuration of heterochromatin and nucleus becomes loose and unstable in aging and the damage it can cause. So cell polarity plays an important role in maintaining that tight configuration.
https://journals.biologists.com/jcs/article/121/8/1141/30524/Cell-polarity-and-cancer-cell-and-tissue-polarity As this paper adds to new evidence that restoration of cell polarity may differentiate the dedifferentiated cancer cells and normalize them. As we age we lose proteins that support bioelectrical
generation and cell polarity. This leads to the fading of both and further leading to eventual loss of the cell.
Cell transcription speed increases with age and leads to more errors in transcription
https://www.nature.com/articles/s41586-023-05922-y
A very interesting paper by Andreas Beyer et. al. Shows us that Transcription of genes is fast but sloppy with age. Six research groups from the University of Cologne Cluster of Excellence on Cellular Stress Responses in Age-Associated Diseases (CECAD), the Max Planck Institute for Biology of Aging (MPI) in Cologne and the University of Göttingen discovered a new molecular mechanism that contributes to ageing by studying the transcription process in five different model organisms and in a wide variety of tissues. 26 scientists investigated genome-wide, age-related changes in transcription processes in nematodes, fruit flies, mice, rats and humans, including diverse tissues. And they discovered that the average speed at which the transcript grows through the attachment of RNA building blocks, the nucleotides, increased with age in all five species. With this increase in the speed of transcription they also saw increase in transcriptional and splicing errors. They observed that increase in transcription speed led to decrease in unspliced exons and intron retention. Basically increased splicing and splicing errors were seen with aging. In my previous blog post Headwaters I have written about how transcription is the beginning of the cascade of biological changes through out our life. So errors in transcription and splicing leads to multiple missexpression of genes which would further lead to cell, tissue and organs becoming impaired and diseased as we see with progressive aging. The authors also found that calorie restriction and inhibition of IGF signaling led to slowing down of transcription speed and corresponding reduction in errors. This also extended their lifespan. Another finding shared by the authors was that overexpression of histones led to reduction transcription speed and increase in lifespan. Also the genes affected were not completely random, as they observed consistent changes across replicates for a subset of introns. As the speed increase in transcription and rise in errors in splicing are global and occur in all
the 5 species including humans studied by the authors and they are not random one can conclude that these age related changes are encoded in our DNA deliberately causing the biological deterioration we see during aging.
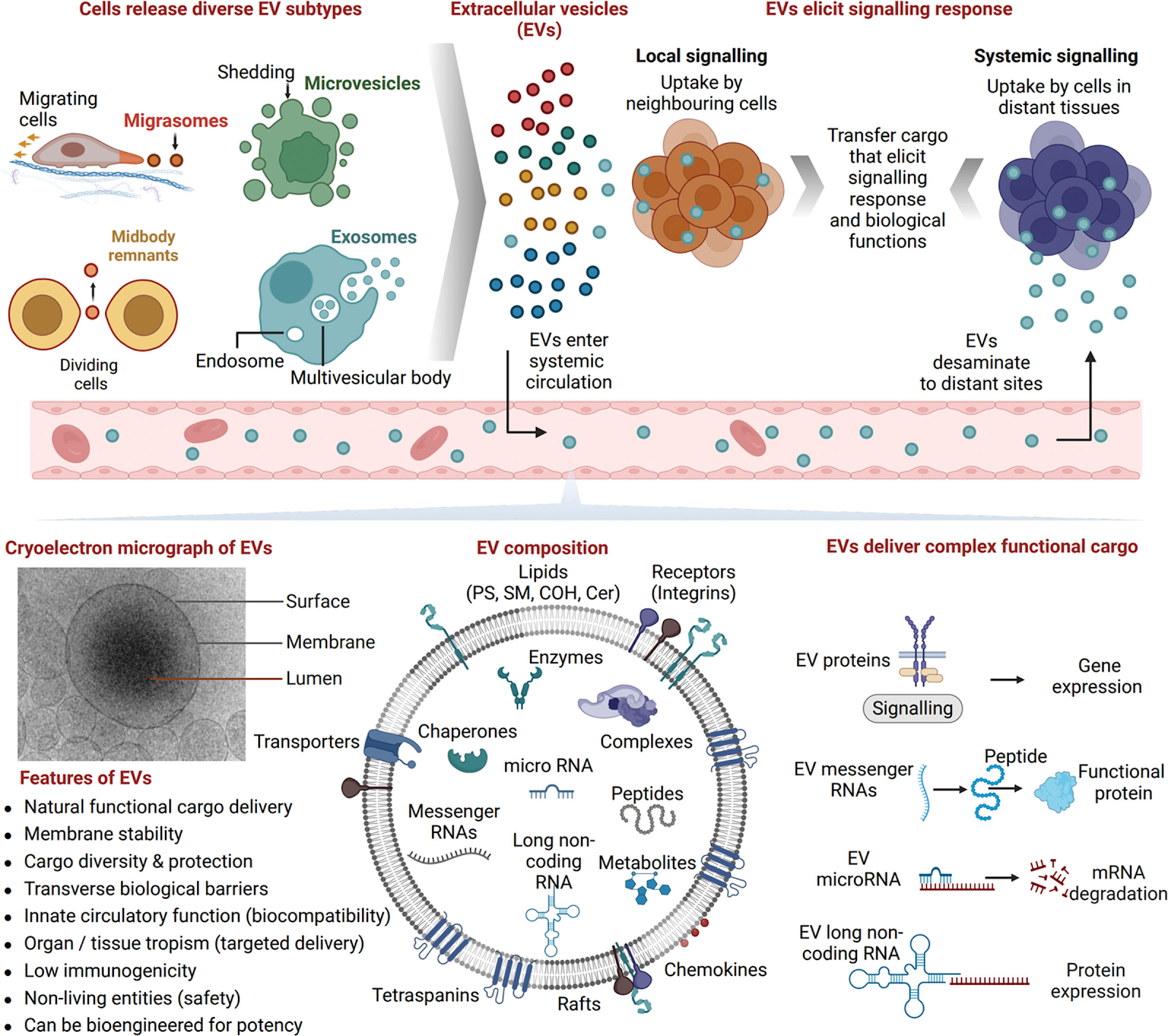
Changes in plasma extracellular vesicles, volume and content, with aging
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5430958/
Cells release lipid-bound extracellular vesicles (EVs; exosomes, microvesicles and apoptotic bodies) containing proteins, lipids and RNAs into the circulation. Vesicles mediate intercellular communication between both neighboring and distant cells. Here the authors analyzed circulating plasma EVs in a cross-sectional and longitudinal study in order to address age-related changes in circulating EVs. They found that EV concentration decreases with advancing age. They also observed increased internalization of EVs by B cells which are immune cells. They found more tumorigenous surface proteins and cargo with age and also more misfolded proteins as cargo. Loss of proteostasis is also cited as a reason by the authors for the reduction of circulating secretome in aging. There are some papers that mention the opposite by showing how EVs increase with age. They cite increase in senescent cells and their EVs increasing the overall volume. But these are harmful EVs and carry cargo that leads to turning an healthy cell into a senescent cell a little like Zombies. That may not change the fact that secretome from healthy cells may decrease with age.
Another paper:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7811845/
Sort of corroborates this. The same authors of the paper cited above Eitan et. al. state in this paper that
They examined whether mtDNA (mitochondrial DNA) can be detected in human plasma EVs and whether mtDNA levels are altered with human age. They found that Plasma EV mtDNA was significantly and negatively associated with age in their cross‐sectional analyses. Basically mtDNA in EVs reduced with age. Did this have any consequences? To determine whether age‐related changes in plasma EV mtDNA affect mitochondrial function, they pooled EV populations of young and old EVs and measured the cell’s respiration rate. They found that cells treated with young EVs had significantly higher levels of both basal and maximal respiration compared to those cells treated with old EVs. So the reduction of mtDNA in EVs in aging did hamper mitochondrial functions.
Longer transcripts decrease with age and shorter transcripts increase with age
https://www.nature.com/articles/s43587-022-00317-6
This has been one of the most interesting papers recently in the aging research field. Not only did they find that longer transcripts seem to fade away as we age and shorter transcripts seem to be of higher abundance but they also found the correlation of longest transcripts with pro-longevity genes and shortest transcripts with genes reported to shorten life.
https://www.nature.com/articles/s41588-022-01279-6
This paper by Joris Pothof et.al. also finds in old mice (2year old) liver cells that there is 40% drop in transcription. This change was pronounced especially in longer transcripts. They did not find any changes or drops due to drop in promoter activity or drop in RNAP II activity. Even the launch of transcription was unaffected. They postulate that DNA breaks interrupted and stalled transcription – and as longer genes/transcripts have greater chances of being broken somewhere they can be the primary cause of their reduced transcription during aging. If we see the first listed change in aging in this post it is talking about rising Double Strand DNA breaks due to cell-free chromatin shards from dead cells which explains why the authors found a 40% drop in transcription. This may also explain what we read above where transcription errors increase with age. Breaks in DNA could stall transcription or create an error in transcription.
The role of retrotransposable elements in aging
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8600649/
There is an on going war from 100s of thousands of years within us. It’s not just against outside pathogens and parasites but also against parasites that have virus like capabilities that have inserted in our genome – not just our genome but it seems in all living life forms-in a big way. These foreign invaders that have made our genome their permanent home since the dawn of life are called Transposons. Scarily they also have the ability to fly from one genomic location to another and insert themselves there. Transposons belong to two main groups: those that move using a DNA intermediate (‘DNA transposons’) in a ‘cut and paste’ mechanism, and retrotransposable elements (retrotransposons) that move using a ‘copy and paste’ mechanism that involves an RNA intermediate. Thirty five percent of the human genome is comprised of retrotransposon DNA sequence! Retrotransposons are further divided into Long Terminal Repeat (LTR) elements, derived from exogenous retrovirus infections, and more primitive and ancient non-LTR elements with an obligate intra-cellular life cycle. Non-LTR retrotransposons consist of two main groups: the Long INnterspersed Elements (LINEs), which encode their own proteins necessary for retrotransposition, and the Short INnterspersed Elements (SINEs), which are short, non-coding RNAs that hijack the LINE protein machinery. Our original genome continues to build tools and mechanisms to silence these elements and the transposons keep trying to outrun them. Both try to outsmart each other but also coexist and adapt from each other. A retrotransposon onslaught can be profoundly deleterious and hence the germline is guarded with multiple defenses. Host defenses are highly effective, and hence the majority of retrotransposons in our genome are passive passengers, slowly accumulating mutations, deletions or other rearrangements. While some older elements can still affect host function through cis-acting gene regulatory or recombinational mechanisms, the deleterious effects of retrotransposons increasingly being linked with aging appear to be largely dependent on the activities of their encoded proteins, and fall into three general mechanisms: genetic and epigenetic effects associated with retrotransposition, DNA damage associated with active or abortive retrotransposition, and activation of immune pathways associated with detection of retrotransposon nucleic acids. One of the key defense mechanisms is heterochromatization which represses these elements. The other retrotransposon silencing mechanism is through methylation. Our genome has proteins that activate these repressive mechanisms on transposons. One example is KRAB domain containing Zinc Finger proteins or KZFPs. KZFPs, of which there are over 400 in the human genome, co-evolve with retrotransposons as part of an ongoing ‘arms race’. Many of these KZFPs bind to retrotransposon elements in the genome and promote their heterochromatinization via the recruitment of KAP1. Retrotransposons mutate to avoid this surveillance, and the host organisms evolve variant KZFPs that can again repress the new generation of retrotransposons. Other such transposon surveillance and repression elements are siRNAs and piRNAs, we have CRISPR like RNA editing enzymes like APOBEC and ADAR, MOV10, BRCA1, SAMHD1 and TREX1.
Some of these factors are defenses against exogenous viruses and act by diverse mechanisms such as editing of viral/retrotransposon genomes to mis-code the protein sequences (APOBEC), decreasing nucleotide triphosphate pools to limit viral/retrotransposon cDNA synthesis (SAMHD1) or degrading viral/retrotransposon nucleic acids (TREX1). These are our intelligent weapons to watch over and overpower transposons. As we read above aging is associated with loss of heterochromatin in various locations in the genome. Decreases of heterochromatin or heterochromatin-establishing factors contribute to elevated retrotransposon activity with age. Genetic interventions that promote heterochromatin formation and/or retrotransposon silencing, remarkably, can increase life span. Aging also sees loss of methylation which also derepresses transposons. Mobilisation of transposons can lead to genomic instability: if a transposon jumps into a functional (coding or regulatory) region of the genome, the insertion often results in loss of function, thereby, facilitating the death of the affected cell. The mass occurrence of transposition events can lead to various degenerative processes. Unwanted activation of transposons in aging can lead to diseases even cancer. Their activation also led to immune response which is part of rise of chronic inflammation seen in aging. Studies suggest that retrotransposons causally contribute to the aging process, and that interventions that oppose retrotransposon activity might improve healthy longevity and lifespan. To quote the authors further: The most proximal approach to inhibiting retrotransposons would be to strengthen the epigenetic mechanisms responsible for their silencing, especially those that become compromised with age. One such epigenetic regulator is SIRT6. Male mice overexpressing SIRT6 show increased life span, which may be due in part to more efficient silencing of L1 elements and reduced inflammation. SIRT6 is involved in multiple processes which include DNA repair, telomere maintenance and metabolism. Small molecule activators of SIRT6 are being developed and may provide an array of health benefits, including improved retrotransposon silencing. Several experiments also show that that a key intervention already known to increase lifespan, a low-calorie diet, dramatically delays the onset of increased transposon activity.

The decline of the extracellular matrix begins just after puberty
https://karger.com/ger/article/66/3/266/148307/The-Matrisome-during-Aging-and-Longevity-A-Systems
We assume that ECM begins to deteriorate due to accumulation of AGE’s and cross linking of collagen over decades but actually ECM is deliberately hurtled towards destruction much earlier: During development and growth, cells constantly remodel the ECM by degrading parts of their ECM and through de novo synthesis of matrisome components in order to maintain homeostasis. This dynamic and energy-intensive process declines after reproduction. Either because natural selection, as defined as reproductive fitness, is ineffective after reproduction, or because of a shift in resource allocation from somatic to germline tissue during the onset of reproduction. Irrespective of the etiology, this predicts that after reproduction the homeostasis of matrisome components, that is, de novo synthesis and ECM remodeling, would decline and should be reflected in the temporal change of matreotypes during aging. Thus, the matreotype of a young ECM is different compared to an old ECM. In this paper the authors also give an explanation of the kind vicious cycle such early decline would lead to: During aging, either through collagen fragmentation or loss of adherence proteins, cells detach from the ECM potentially leading to cell dysfunction and loss of ECM synthesis and turnover. In fact, the loss of ECM-to-cell connection might start a vicious downward spiral. For instance, during aging, there is an increase in activity of ECM-degrading enzymes, such as matrix metalloproteases (MMP). Increased MMP activity leads to collagen fragmentation, causing cell detachment, which leads to altered -integrin signaling and an increase in mitochondrial -reactive oxygen species, which in turn promotes the -expression of more MMPs, leading to further ECM fragmentation.
So the aging program seems to select our most important components and systems for early start in deterioration -another example is 60% loss of heat shock protein chaperone support to protein product also happening just after puberty. ECM is as important as protein production because it makes up a staggering 50–70% of the human body mass and any change in that would be felt systemically. Another way to estimate importance of decline of ECM in aging is from this paper:
https://www.aginganddisease.org/EN/10.14336/AD.2022.1116
The authors found that 12 most established longevity-promoting transcription factors (i.e., CREB1, FOXO1,3, GATA1,2,3,4, HIF1A, JUN, KLF4, MYC, NFE2L2/Nrf2, RELA/NF-κB, REST, STAT3,5A, and TP53/p53), directly and indirectly transcriptionally regulate ECM genes.
Healthy cell-ECM crosstalk is vital for cellular homeostasis
Cells synthesize their own surrounding ECM. Each type of cell has its own unique type of EVM. Throughout the life of a cell there is constant cross talk with its ECM-in fact during aging when the damage to ECM is so great that a cell detached from it, which is called anoikis leading to the cell either dying or turning cancerous. A fascinating finding is that placing senescent cells or aged stem cells in a “younger ECM” has been shown to rejuvenate these old cells. The ECM provides instructive signals that change cellular function and identity. For instance, placing tumor cells into an embryonic ECM reprograms them to lose their tumorigenicity and become normal cells. Furthermore, the lost regenerative potential of old muscles is rejuvenated by grafting them into young, but not old hosts. And so a young and healthy ECM is vital for the survival of the cell it surrounds. During aging, components of the ECM become damaged through fragmentation, glycation, crosslinking, and accumulation of protein aggregation, all of which contribute to age-related pathologies. Since ECM is pervasive throughout our body it’s decline is linked to catastrophic tissue failure and multiple organ failure. ECM is a dynamic structure that is continuously built and remodeled but in aging Advanced Glycation End products that build up on the ECM blocking remodeling enzymes making it stiff. This leads to diseases like fibrosis seen in various organs like heart, lungs, kidneys, etc. So far it was believed that once formed AGEs can not be destroyed. But in 2020 Nam Y Kim et.al. published this paper: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6768434/
In which they shared about an altered enzyme and it’s variant were able to reverse AGEs. This is a great breakthrough and hopefully all types of AGEs can soon be eliminated.
These are just a few of the thousands of changes occurring during aging. From this we can see why addressing one or a few of these changes will not be able to reverse aging. Only those interventions that work to reverse all these changes back to normal or youthful configuration can actually reverse aging. The entire universe is made up of only one building material: atoms. Atoms are incredibly mysterious full of magical forces but atomic, nuclear and quantum physics deserve a separate blog post. What is fascinating is what happened on Earth: these mystical atoms in different configurations formed molecules. From these molecules came living forms and their unit is cells. Cells are intricately beautiful with their components and engineering. Along with other tiny life forms like microbes and and another mysterious object: virus-it’s like a non-living zombie code that can jump from living cells to living cells housed in various bodies. Various collections have developed of these three to form a variety of life forms including us humans. Another principle of the universe seen in everything, living or inanimate, is recycling. May fly lives just 24 hours and our Sun 4.5 billion years but despite that difference both have one thing in common: both will perish and be recycled into something else. And there is black holes from which nothing seems to escape: the ultimate recycler of the known universe. On Earth cells have DNA and RNA which is DNA’s master and servant. But emerging evidence over the last few decades points to a recycling code embedded in the DNA which is altered due to various paths of adaptationary changes adopted by each life form or each unique collection of cells, microbes and viruses. No matter what alterations almost all seem to participate in the universal principle of recycling. In some life forms reproduction is asexual and automatic as part of their life journey. In other forms with higher intelligence where a factor of choice comes in reproduction is incentivized with pleasurable sensations. So the birth and death cycle continues millions of times. In life forms with even more complex engineering, like humans, we can witness an army of agents of time which chip away at a thousand different locations till one day the life form succumbs and dies. Our awareness of our biology is growing rapidly and every now and then we keep discovering another agent of time. Our gift from adaptions to survive is highest intelligence amongst all life forms on Earth due to which we have come to totally dominate Earth just like our predecessors the dinosaurs. This intelligence will also allow us at some point to synthesize our own adaptation to overpower these agents of time. We have the potential to become one of the first forms in the universe to bypass the principle of recycling at least from our own short recycling cycles. Although eventually one of the powerful forces of the universe would end our run. If we reach the point of immortality who knows we may get enough time to avoid or prevent total oblivion through interstellar travel and building our own defenses manipulating matter. Becoming biologically immortal is not such a big jump as we think now because as I have said in my previous posts there are complex life forms on Earth not gifted with our intelligence that have already achieved this by way of adaptation lottery win. So our intelligence will surely allow us to create indefinite lifespan in perpetual youthful homeostasis. We are going to build technologies which we can’t even imagine today. Tempted to throw in just one example of possibility: The virus that recently created a global epidemic is considered an enemy but we have the capability to turn an enemy into our greatest weapon. One song that all agents of time sing is dialing down of our amazing repair systems so imagine tiny nano robotized virus like codes that continuously repair damage as it occurs in our 30 trillion cells and their microenvironment. We can literally blow this virus from various points in a city to ensure everyone remains infected. The virus can be engineered to sit in a cell and get activated at any sign of damage and repair it. Similar virus can also be created to prevent or treat cancer our other great enemy. May be we can learn from cancer cell and reengineer it to make us young – after all a cancer cell makes itself immortal. It would be technologies we can’t even begin to comprehend now.